 |
JDFTx
1.7.0
|


|
|
JDFTx
1.7.0
|
|
All the calculations so far have been of isolated molecules in vacuum, which is a good description for dilute gases. However, chemical reactions often occur in a solution, and predicting reaction mechanisms using DFT requires calculating structures and free energies of molecules in solution. Directly describing the solvent using its constituent molecules requires several thousands of DFT calculations to sample all configurations of solvent molecules, and is extremely computationally intensive. A practical alternative is to use a continuum solvation model which directly approximates the equilibrium effect of the solvent on a solute, without treating individual solvent configurations.
A major focus in the development of JDFTx has been the development of a heirarchy of solvation models from continuum to a detailed classical DFT approach, within the framework of Joint Density Functional Theory. (That's where JDFTx gets its J!) This tutorial shows how to put molecules in solution and introduces some of the commands that control solvation and fluid properties.
Let's put a water molecule in liquid water. The free energy of this process is related to the vapor pressure of liquid water and can be calculated from experimental measurements as -0.0101 Hartrees (= -6.33 kcal/mol). First, we perform the calculation in vacuum using the input files
#Save to common.in lattice Cubic 15 coulomb-interaction Isolated coulomb-truncation-embed 0 0 0 ion-species GBRV/$ID_pbe.uspp elec-cutoff 20 100 coords-type cartesian
containing the shared commands as before, and
#Save to Vacuum.in and run as jdftx -i Vacuum.in | tee Vacuum.out: include common.in ion O 0.00 0.00 0.00 0 ion H 0.00 1.13 +1.45 1 ion H 0.00 1.13 -1.45 1 ionic-minimize nIterations 10 dump-name Vacuum.$VAR dump End State
Upon completion, this calculation will output the optimized geometry and wavefunctions of a water molecule in vacuum. Next, we run a solvated calculation starting from this state:
#Save to LinearPCM.in and run as jdftx -i LinearPCM.in | tee LinearPCM.out: include common.in include Vacuum.ionpos initial-state Vacuum.$VAR ionic-minimize nIterations 10 dump-name LinearPCM.$VAR dump End State BoundCharge fluid LinearPCM #Simple continuum fluid with linear response pcm-variant GLSSA13 #default, if not specified fluid-solvent H2O #Solvent is water
This runs a solvated calculation with the simplest GLSSA13 LinearPCM model (this is exactly the same model which was later implemented in VASP as VASPsol) and outputs the bound charge density induced in the solvent by electrostatic interactions with the solute. Examine the output file: now there are lines starting with 'Linear fluid' between electronic steps. These correspond to optimization of the fluid degrees of freedom (in this simple case, by solving a linearized Poisson-Boltzmann equation) in response to the solute's charge density. The energy components now include an additional piece Adiel, which is the total contribution due to the solvent.
The solvation free energy is the difference between the final energies in Vacuum.out and LinearPCM.out, which turns out to be (-17.2682) - (-17.2795) = -0.0113 Hartrees in good agreement with the experimental estimate of -0.0101 Hartrees. Visualizing the bound charge density (nbound) using
createXSF LinearPCM.out LinearPCM.xsf nbound
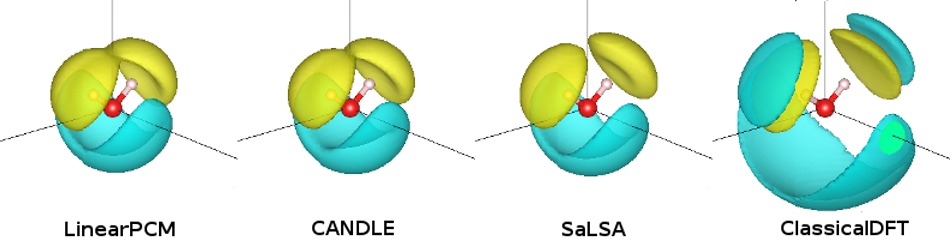
results in the first panel of the figure above.
The command fluid selects the broad class of solvation model, some of which support many variants selected by command pcm-variant. Some of the common / recommended models are:
fluid LinearPCM pcm-variant GLSSA13
fluid LinearPCM pcm-variant SCCS_*
fluid LinearPCM pcm-variant CANDLE
fluid NonlinearPCM pcm-variant GLSSA13
fluid SaLSA
fluid ClassicalDFTAlso see the various commands listed under fluid in the Input file documentation page for controlling numerous details of the fluid calculation.
Now, as an exercise run the water calculation with each class of solvation model, compare the solvation energies and visualize the bound charge in the liquid using createXSF and VESTA, as shown below and at the start of this tutorial. (SaLSA and ClassicalDFT will take a while!) Note how LinearPCM, the nonlocally-inspired variant CANDLE, as well as the non-local model SaLSA have the bound charge mostly restricted to a surface surrounding the solute, whereas ClassicalDFT, exhibits a shell structure with several layers surrounding the solute. Additionally, in this simple case of a neutral solute, all the solvation models are comparably accurate.